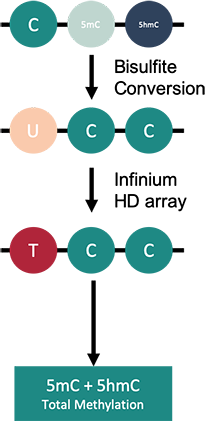
How to properly cite our product/service in your work We strongly recommend using this: Infinium MethylationEPIC Array v2.0 Service (Hologic Diagenode Cat# G02090006). Click here to copy to clipboard. Using our products or services in your publication? Let us know! |
Molecular profiling of chemotherapy-resistant breast cancer reveals DNA methylation remodeling associated with the acquisition of paclitaxel resistance
Trnkova, Lenka et. al.
Aims
Chemotherapy resistance remains a major challenge in breast cancer (BC) treatment. This study aimed to investigate the role of DNA methylation in this complex process and evaluate the potential of the DNA methyltransferase inhibitor decitabine (DAC) in restoring chemosensitivity.
Methods
Paclitaxel (... |
The Peripheral Epigenome Predicts White Matter Volume Contingent on Developmental Stage: An ECHO Study
Hanson, T. et al.
White matter (WM) development is crucial for efficient neural connectivity, and disruptions in its maturation are associated with neurocognitive and psychiatric disorders. Emerging evidence suggests that epigenetic mechanisms, particularly DNA methylation (DNAm), regulate WM structure, yet little is known about thes... |
Effects of Maternal Peanut Intake and Breastfeeding Duration on Offspring DNA Methylation
Garay, Jessica L et al.
Emerging evidence suggests that maternal diet during pregnancy and breastfeeding may influence epigenetic modifications in offspring genes related to neurodevelopment and inflammation, although the specific mechanisms and long‐term implications of these effects are not well understood. This study evaluated the influ... |
Cell-specific epigenome-wide DNA methylation in peripheral CD4(+) lymphocytes from patients with primary biliary cholangitis
Arvaniti, Pinelopi et al.
Background/aims
Primary biliary cholangitis (PBC) is a chronic cholestatic autoimmune liver disease, triggered by a complex interplay between genetic, environmental and epigenetic factors. We investigated the methylation profile of peripheral CD4(+) lymphocytes from PBC patients compared to healthy controls (HC) an... |
Comprehensive molecular portrait reveals genetic diversity and distinct molecular subtypes of small intestinal neuroendocrine tumors
Céline Patte et al.
Small intestinal neuroendocrine tumors (siNETs) are rare bowel tumors arising from malignant enteroendocrine cells, which normally regulate digestion throughout the intestine. Though infrequent, their incidence is rising through better diagnosis, fostering research into their origin and treatment. To date, siNETs ar... |
Clinical Value of MLPA for Prognostic Assessment of Chromosomal Rearrangements and DNA Methylation in Uveal Melanoma
Andrea Soltysova et al.
Purpose: Uveal melanoma (UM) is the most prevalent primary intraocular malignancy in adults, with prognosis significantly influenced by genetic and epigenetic factors. Reliable and cost-effective methods to detect chromosomal aberrations and DNA methylation changes are essential for improving prognostication an... |
Using multiomic integration to improve blood biomarkers of major depressive disorder: a case-control study
Amazigh Mokhtari et al.
Background
Major depressive disorder (MDD) is a leading cause of disability, with a twofold increase in prevalence in women compared to men. Over the last few years, identifying molecular biomarkers of MDD has proven challenging, reflecting interactions among multiple environmental and genetic factors. Recently, ... |
Genome-wide transcriptome and DNA methylome profiling of acquired cystic disease-associated renal cell carcinoma
Hiroki Ishihara et al.
Acquired cystic disease (ACD)-associated renal cell carcinoma (RCC) develops uniquely and frequently in patients receiving long-term dialysis for end-stage renal disease (ESRD). In our previous study, the molecular alteration profiles of ACD-associated RCC were partially similar to those of papillary RCC (PRCC). How... |
Accelerated epigenetic ageing after burn injury
Jack Sullivan et al.
Individuals who suffer a major burn injury are at higher risk of developing a range of age-associated diseases prematurely leading to an increase in mortality in adult and juvenile burn injury survivors. One possible explanation is that injury is accelerating the biological ageing process. To test this hypothesis, w... |
Concurrent RB1 and P53 pathway disruption predisposes to the development of a primitive neuronal component in high-grade gliomas depending on MYC-driven EBF3 transcription
Francesca Pagani et al.
The foremost feature of glioblastoma (GBM), the most frequent malignant brain tumours in adults, is a remarkable degree of intra- and inter-tumour heterogeneity reflecting the coexistence within the tumour bulk of different cell populations displaying distinctive genetic and transcriptomic profiles. GBM with primiti... |
Epigenetic Analysis of ST3GAL3 and other Sialic Acid Metabolism Genes in ADHD
Lillian Dipnall et al.
Research indicates that the underlying neurobiology of Attention Deficit/Hyperactivity Disorder (ADHD) may stem from a combination of genetic and environmental contributions. Genetic and epigenetic research have highlighted the potential role of the sialtransferase gene ST3GAL3 in this process. Adopting a ... |
Matched analysis of detailed peripheral blood and tumor immune microenvironment profiles in bladder cancer
Chen JQ et al.
Background: Bladder cancer and therapy responses hinge on immune profiles in the tumor microenvironment (TME) and blood, yet studies linking tumor-infiltrating immune cells to peripheral immune profiles are limited. Methods: DNA methylation cytometry quantified TME and matched peripheral blood immune ... |
Altered DNA methylation and gene expression predict disease severity inpatients with Aicardi-Goutières syndrome.
Garau J. et al.
Aicardi-Goutières Syndrome (AGS) is a rare neuro-inflammatory disease characterized by increased expression of interferon-stimulated genes (ISGs). Disease-causing mutations are present in genes associated with innate antiviral responses. Disease presentation and severity vary, even between patients with ident... |
DNA methylation aberrancy is a reliable prognostic tool in uveal melanoma
Soltysova A. et al.
Despite outstanding advances in understanding the genetic background of uveal melanoma (UM) development and prognosis, the role of DNA methylation reprogramming remains elusive. This study aims to clarify the extent of DNA methylation deregulation in the context of gene expression changes and its utility as a reliab... |
Decitabine increases neoantigen and cancer testis antigen expression toenhance T cell-mediated toxicity against glioblastoma.
Ma Ruichong et al.
BACKGROUND: Glioblastoma (GBM) is the most common and malignant primary brain tumour in adults. Despite maximal treatment, median survival remains dismal at 14-24 months. Immunotherapies, such as checkpoint inhibition, have revolutionised management of some cancers but have little benefit for GBM patients. This is, ... |
Enhanced cell deconvolution of peripheral blood using DNA methylation for high-resolution immune profiling
Lucas A Salas, Ze Zhang, Devin C Koestler, Rondi A Butler, Helen M Hansen, Annette M Molinaro, John K Wiencke, Karl T Kelsey, Brock C Christensen
DNA methylation microarrays can be employed to interrogate cell-type composition in complex tissues. Here, we expand reference-based deconvolution of blood DNA methylation to include 12 leukocyte subtypes (neutrophils, eosinophils, basophils, monocytes, B cells, CD4+ and CD8+ naïve and memory cells, natural kil... |
Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders
Guillaume Velasco, Damien Ulveling,Sophie Rondeau,Pauline Marzin,Motoko Unoki,Valérie Cormier-Daire, Claire Francastel
DNA methylation (DNAme) profiling is used to establish specific biomarkers to improve the diagnosis of patients with inherited neurodevelopmental disorders and to guide mutation screening. In the specific case of mendelian disorders of the epigenetic machinery, it also provides the basis to infer mechanistic aspects... |
Genome-wide DNA methylation and transcriptome integration reveal distinct sex differences in skeletal muscle
Shanie Landen, Macsue Jacques , Danielle Hiam , Javier Alvarez, Nicholas R Harvey, Larisa M. Haupt, Lyn R, Griffiths, Kevin J Ashton, Séverine Lamon, Sarah Voisin, Nir Eynon
Nearly all human complex traits and diseases exhibit some degree of sex differences, and epigenetics contributes to these differences as DNA methylation shows sex differences in various tissues. However, skeletal muscle epigenetic sex differences remain largely unexplored, yet skeletal muscle displays distinct sex d... |
ZNF718, HOXA4, and ZFP57 are differentially methylated inperiodontitis in comparison with periodontal health: Epigenome-wide DNAmethylation pilot study.
Hernández H.G. et al.
OBJECTIVE: To investigate the differences in the epigenomic patterns of DNA methylation in peripheral leukocytes between patients with periodontitis and gingivally healthy controls evaluating its functional meaning by functional enrichment analysis. BACKGROUND: The DNA methylation profiling of peripheral leukocytes ... |
From methylation to myelination: epigenomic and transcriptomic profilingof chronic inactive demyelinated multiple sclerosis lesions
Tiane A. et al.
Introduction In the progressive phase of multiple sclerosis (MS), the hampered differentiation capacity of oligodendrocyte precursor cells (OPCs) eventually results in remyelination failure. We have previously shown that DNA methylation of Id2/Id4 is highly involved in OPC differentiation and remyelination. In this ... |