How to properly cite our product/service in your workWe strongly recommend using this: RNA Data Analysis (Hologic Diagenode Cat# G02030005). Click here to copy to clipboard. Using our products or services in your publication? Let us know! |
MGcount: a total RNA-seq quantification tool to address multi-mappingand multi-overlapping alignments ambiguity in non-coding transcripts |
RNA Data Analysis
Catalog Number
Format
G02030005
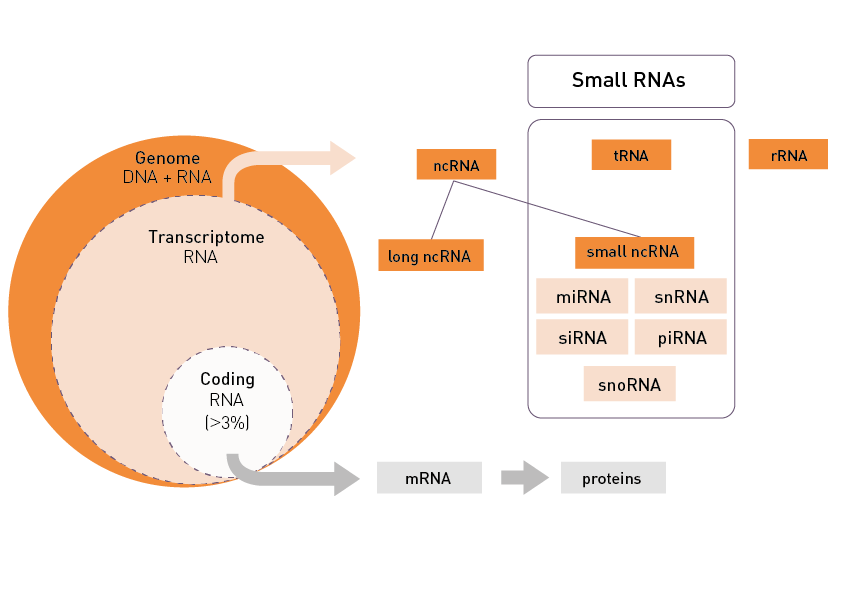
Total RNA sequencing (RNA-Seq) detects both coding and noncoding RNAs and is typically used to measure gene and transcript abundance as well as to identify novel components of the transcriptome. Messenger RNA-Seq focuses on the quantification of gene expression, the identification of unknown transcripts, the discovery of alternative splicing and gene fusion events. And finally, small non-coding RNA sequencing (sncRNA-Seq) will detect small (<100 nucleotides long) RNAs that operate as key regulators in cellular processes.
What do we provide with the analysis?
This analysis provides information for either genes or isoforms with their expression levels.
Standard Analysis
- Summary statistics (total sequenced reads, total mapping reads, uniquely aligned reads, PCR duplicates, number of genes detected, average read density per gene, number of highly expressed genes, etc.)
- Trimmed and filtered reads in fastQ files after sequencing QC
- BAM sorted files from alignment to reference genome or transcriptome (indexed bam files and bigwig files included)
- Matrix with expression abundance obtained with specialized quantification tool MGCount (software developed by Diagenode). A table of MG communities linking each original feature in the GTF file with the resultant count matrix and metadata feature identifiers.
Advanced Analysis
- Comparative analysis (also called differential analysis) aimed at finding differentially expressed genes (DEGs) between two groups of samples
- Functional gene annotation
- Gene ontology enrichment analysis on DEGs
- Pathway enrichment analysis on DEGs (KEGG or DOSE for human samples)
- Alternative splicing analysis
- Gene fusion analysis
- Novel transcript identification
Customized Analysis
If you require a type of analysis that is not in the previous list, please consult with our expert bioinformatics team.