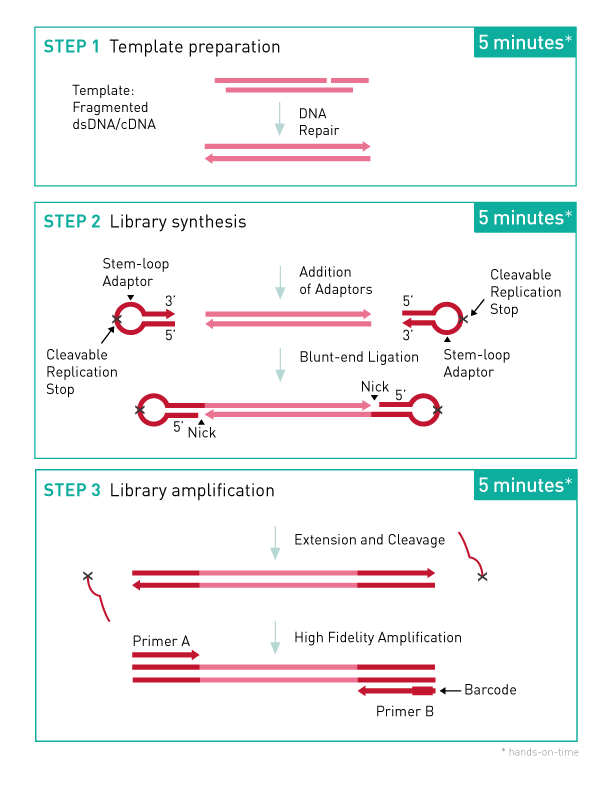
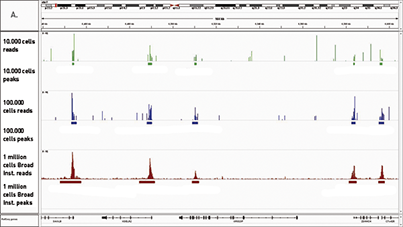
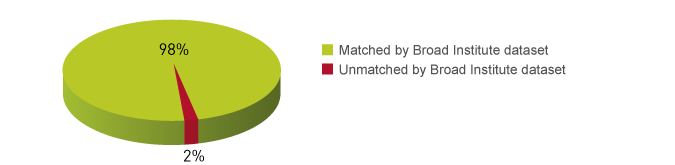
How to properly cite our product/service in your work We strongly recommend using this: MicroPlex Library Prep. Kit v2 (12 indexes) - DISCONTINUED (Hologic Diagenode Cat# C05010012). Click here to copy to clipboard. Using our products or services in your publication? Let us know! |
Differentiation block in acute myeloid leukemia regulated by intronicsequences of FTO
Camera F. et al.
Iroquois transcription factor gene IRX3 is highly expressed in 20–30\% of acute myeloid leukemia (AML) and contributes to the pathognomonic differentiation block. Intron 8 FTO sequences ∼220kB downstream of IRX3 exhibit histone acetylation, DNA methylation, and contacts with th... |
Supraphysiological Androgens Promote the Tumor Suppressive Activity of the Androgen Receptor Through cMYC Repression and Recruitment of the DREAM Complex
Nyquist M. et al.
The androgen receptor (AR) pathway regulates key cell survival programs in prostate epithelium. The AR represents a near-universal driver and therapeutic vulnerability in metastatic prostate cancer, and targeting AR has a remarkable therapeutic index. Though most approaches directed toward AR focus on inhibiting AR ... |
In skeletal muscle and neural crest cells, SMCHD1 regulates biologicalpathways relevant for Bosma syndrome and facioscapulohumeral dystrophyphenotype.
Laberthonnière C. et al.
Many genetic syndromes are linked to mutations in genes encoding factors that guide chromatin organization. Among them, several distinct rare genetic diseases are linked to mutations in SMCHD1 that encodes the structural maintenance of chromosomes flexible hinge domain containing 1 chromatin-associated factor. In hu... |
Hypomethylation and overexpression of Th17-associated genes is ahallmark of intestinal CD4+ lymphocytes in Crohn's disease.
Sun Z. et al.
BACKGROUND: The development of Crohn's disease (CD) involves immune cell signaling pathways regulated by epigenetic modifications. Aberrant DNA methylation has been identified in peripheral blood and bulk intestinal tissue from CD patients. However, the DNA methylome of disease-associated intestinal CD4 + lymphocyte... |
Mutant FUS induces chromatin reorganization in the hippocampus andalters memory processes.
Tzeplaeff L. et al.
Cytoplasmic mislocalization of the nuclear Fused in Sarcoma (FUS) protein is associated to amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Cytoplasmic FUS accumulation is recapitulated in the frontal cortex and spinal cord of heterozygous Fus mice. Yet, the mechanisms linking FUS mislocalizati... |
Transient suppression of SUMOylation in embryonic stem cells generatesembryo-like structures.
Cossec J-C. et al.
Recent advances in synthetic embryology have opened new avenues for understanding the complex events controlling mammalian peri-implantation development. Here, we show that mouse embryonic stem cells (ESCs) solely exposed to chemical inhibition of SUMOylation generate embryo-like structures comprising anterior neura... |
ZEB1 controls a lineage-specific transcriptional program essential formelanoma cell state transitions
Tang Y. et al.
Cell plasticity sustains intra-tumor heterogeneity and treatment resistance in melanoma. Deciphering the transcriptional mechanisms governing reversible phenotypic transitions between proliferative/differentiated and invasive/stem-like states is required in order to design novel therapeutic strategies. EMT-inducing ... |
Gene Regulatory Interactions at Lamina-Associated Domains
Madsen-Østerbye J. et al.
The nuclear lamina provides a repressive chromatin environment at the nuclear periphery. However, whereas most genes in lamina-associated domains (LADs) are inactive, over ten percent reside in local euchromatic contexts and are expressed. How these genes are regulated and whether they are able to interact with regu... |
Mechanisms and function of de novo DNA methylation in placentaldevelopment reveals an essential role for DNMT3B.
Andrews S. et al.
DNA methylation is a repressive epigenetic modification that is essential for development, exemplified by the embryonic and perinatal lethality observed in mice lacking de novo DNA methyltransferases (DNMTs). Here we characterise the role for DNMT3A, 3B and 3L in gene regulation and development of the mouse placenta... |
TCDD induces multigenerational alterations in the expression ofmicroRNA in the thymus through epigenetic modifications
Singh Narendra P et al.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), a potent AhR ligand, is an environmental contaminant that is known for mediating toxicity across generations. However, whether TCDD can induce multigenerational changes in the expression of miRNAs (miRs) has not been previously studied. In the current study, we investigate... |
The histone acetyltransferase KAT6A is recruited to unmethylatedCpG islands via a DNA binding winged helix domain.
Weber L.M. et al.
The lysine acetyltransferase KAT6A (MOZ, MYST3) belongs to the MYST family of chromatin regulators, facilitating histone acetylation. Dysregulation of KAT6A has been implicated in developmental syndromes and the onset of acute myeloid leukemia (AML). Previous work suggests that KAT6A is recruited to its genomic targ... |
Polyglutamine-expanded ATXN7 alters a specific epigenetic signatureunderlying photoreceptor identity gene expression in SCA7 mouseretinopathy.
Niewiadomska-Cimicka A.et al.
BACKGROUND: Spinocerebellar ataxia type 7 (SCA7) is a neurodegenerative disorder that primarily affects the cerebellum and retina. SCA7 is caused by a polyglutamine expansion in the ATXN7 protein, a subunit of the transcriptional coactivator SAGA that acetylates histone H3 to deposit narrow H3K9ac mark at DNA regula... |
Intranasal administration of Acinetobacter lwoffii in a murine model ofasthma induces IL-6-mediated protection associated with cecal microbiotachanges.
Alashkar A. B. et al.
BACKGROUND: Early-life exposure to certain environmental bacteria including Acinetobacter lwoffii (AL) has been implicated in protection from chronic inflammatory diseases including asthma later in life. However, the underlying mechanisms at the immune-microbe interface remain largely unknown. METHODS: The effects o... |
Identification of genomic binding sites and direct target genes for thetranscription factor DDIT3/CHOP.
Osman A. et al.
DDIT3 is a tightly regulated basic leucine zipper (bZIP) transcription factor and key regulator in cellular stress responses. It is involved in a variety of pathological conditions and may cause cell cycle block and apoptosis. It is also implicated in differentiation of some specialized cell types and as an oncogene... |
Cryptococcal Hsf3 controls intramitochondrial ROS homeostasis byregulating the respiratory process.
Gao X.et al.
Mitochondrial quality control prevents accumulation of intramitochondrial-derived reactive oxygen species (mtROS), thereby protecting cells against DNA damage, genome instability, and programmed cell death. However, underlying mechanisms are incompletely understood, particularly in fungal species. Here, we show that... |
Dominant role of DNA methylation over H3K9me3 for IAP silencingin endoderm.
Wang Z. et al.
Silencing of endogenous retroviruses (ERVs) is largely mediated by repressive chromatin modifications H3K9me3 and DNA methylation. On ERVs, these modifications are mainly deposited by the histone methyltransferase Setdb1 and by the maintenance DNA methyltransferase Dnmt1. Knock-out of either Setdb1 or Dnmt1 leads to... |
HDAC1 and PRC2 mediate combinatorial control in SPI1/PU.1-dependentgene repression in murine erythroleukaemia.
Gregoricchio S. et al.
Although originally described as transcriptional activator, SPI1/PU.1, a major player in haematopoiesis whose alterations are associated with haematological malignancies, has the ability to repress transcription. Here, we investigated the mechanisms underlying gene repression in the erythroid lineage, in which SPI1 ... |
Dual role of histone variant H3.3B in spermatogenesis: positiveregulation of piRNA transcription and implication in X-chromosomeinactivation.
Fontaine E. et al.
The histone variant H3.3 is encoded by two distinct genes, H3f3a and H3f3b, exhibiting identical amino-acid sequence. H3.3 is required for spermatogenesis, but the molecular mechanism of its spermatogenic function remains obscure. Here, we have studied the role of each one of H3.3A and H3.3B proteins in spermatogene... |
TBX2 acts as a potent transcriptional silencer of tumour suppressor genesthrough interaction with the CoREST complex to sustain theproliferation of breast cancers.
McIntyre A.J. et al.
Chromosome 17q23 amplification occurs in 20\% of primary breast tumours and is associated with poor outcome. The TBX2 gene is located on 17q23 and is often over-expressed in this breast tumour subset. TBX2 is an anti-senescence gene, promoting cell growth and survival through repression of Tumour Suppressor Genes (T... |
Caffeine intake exerts dual genome-wide effects on hippocampal metabolismand learning-dependent transcription.
Paiva I. et al.
Caffeine is the most widely consumed psychoactive substance in the world. Strikingly, the molecular pathways engaged by its regular consumption remain unclear. We herein addressed the mechanisms associated with habitual (chronic) caffeine consumption in the mouse hippocampus using untargeted orthogonal omics techniq... |
Epigenetic Mechanisms Mediating Cell State Transitions in Chondrocytes
Wuelling M. et al.
Epigenetic modifications play critical roles in regulating cell lineage differentiation, but the epigenetic mechanisms guiding specific differentiation steps within a cell lineage have rarely been investigated. To decipher such mechanisms, we used the defined transition from proliferating (PC) into hypertrophic chon... |
The CpG Island-Binding Protein SAMD1 Contributes to anUnfavorable Gene Signature in HepG2 Hepatocellular CarcinomaCells.
Simon C. et al.
The unmethylated CpG island-binding protein SAMD1 is upregulated in many human cancer types, but its cancer-related role has not yet been investigated. Here, we used the hepatocellular carcinoma cell line HepG2 as a cancer model and investigated the cellular and transcriptional roles of SAMD1 using ChIP-Seq and RNA-... |
Local euchromatin enrichment in lamina-associated domains anticipatestheir repositioning in the adipogenic lineage.
Madsen-Østerbye J. et al.
BACKGROUND: Interactions of chromatin with the nuclear lamina via lamina-associated domains (LADs) confer structural stability to the genome. The dynamics of positioning of LADs during differentiation, and how LADs impinge on developmental gene expression, remains, however, elusive. RESULTS: We examined changes in t... |
CREBBP/EP300 acetyltransferase inhibition disrupts FOXA1-bound enhancers to inhibit the proliferation of ER+ breast cancer cells.
Bommi-Reddy A. et al.
Therapeutic targeting of the estrogen receptor (ER) is a clinically validated approach for estrogen receptor positive breast cancer (ER+ BC), but sustained response is limited by acquired resistance. Targeting the transcriptional coactivators required for estrogen receptor activity represents an alternative approach... |
NuA4 and H2A.Z control environmental responses and autotrophicgrowth in Arabidopsis
Bieluszewski T. et al.
Nucleosomal acetyltransferase of H4 (NuA4) is an essential transcriptional coactivator in eukaryotes, but remains poorly characterized in plants. Here, we describe Arabidopsis homologs of the NuA4 scaffold proteins Enhancer of Polycomb-Like 1 (AtEPL1) and Esa1-Associated Factor 1 (AtEAF1). Loss of AtEAF1 results in ... |
Three classes of epigenomic regulators converge to hyperactivate theessential maternal gene deadhead within a heterochromatin mini-domain.
Torres-Campana D. et al.
The formation of a diploid zygote is a highly complex cellular process that is entirely controlled by maternal gene products stored in the egg cytoplasm. This highly specialized transcriptional program is tightly controlled at the chromatin level in the female germline. As an extreme case in point, the massive and s... |
Epromoters function as a hub to recruit key transcription factorsrequired for the inflammatory response
Santiago-Algarra D. et al.
Gene expression is controlled by the involvement of gene-proximal (promoters) and distal (enhancers) regulatory elements. Our previous results demonstrated that a subset of gene promoters, termed Epromoters, work as bona fide enhancers and regulate distal gene expression. Here, we hypothesized that Epromoters play a... |
Decreased PRC2 activity supports the survival of basal-like breastcancer cells to cytotoxic treatments
Mieczkowska IK et al.
Breast cancer (BC) is the most common cancer occurring in women but also rarely develops in men. Recent advances in early diagnosis and development of targeted therapies have greatly improved the survival rate of BC patients. However, the basal-like BC subtype (BLBC), largely overlapping with the triple-negative BC ... |
Ago1 controls myogenic differentiation by regulating eRNA-mediatedCBP-guided epigenome reprogramming.
Fallatah Bodor et al.
The role of chromatin-associated RNAi components in the nucleus of mammalian cells and in particular in the context of developmental programs remains to be elucidated. Here, we investigate the function of nuclear Argonaute 1 (Ago1) in gene expression regulation during skeletal muscle differentiation. We show that Ag... |
Extensive NEUROG3 occupancy in the human pancreatic endocrine generegulatory network.
Schreiber V. et al.
OBJECTIVE: Mice lacking the bHLH transcription factor (TF) Neurog3 do not form pancreatic islet cells, including insulin-secreting beta cells, the absence of which leads to diabetes. In humans, homozygous mutations of NEUROG3 manifest with neonatal or childhood diabetes. Despite this critical role in islet cell deve... |
Alveolar macrophages from persons living with HIV show impairedepigenetic response to Mycobacterium tuberculosis.
Correa-Macedo Wilian et al.
Persons living with HIV (PLWH) are at increased risk of tuberculosis (TB). HIV-associated TB is often the result of recent infection with Mycobacterium tuberculosis (Mtb) followed by rapid progression to disease. Alveolar macrophages (AM) are the first cells of the innate immune system that engage Mtb, but how HIV a... |
INTS11 regulates hematopoiesis by promoting PRC2 function.
Zhang Peng et al.
INTS11, the catalytic subunit of the Integrator (INT) complex, is crucial for the biogenesis of small nuclear RNAs and enhancer RNAs. However, the role of INTS11 in hematopoietic stem and progenitor cell (HSPC) biology is unknown. Here, we report that INTS11 is required for normal hematopoiesis and hematopoietic-spe... |
The related coactivator complexes SAGA and ATAC control embryonicstem cell self-renewal through acetyltransferase-independent mechanisms
Fischer Veronique et al.
SUMMARY SAGA (Spt-Ada-Gcn5 acetyltransferase) and ATAC (Ada-two-A-containing) are two related coactivator complexes, sharing the same histone acetyltransferase (HAT) subunit. The HAT activities of SAGA and ATAC are required for metazoan development, but the role of these complexes in RNA polymerase II transcription ... |
Altered Chromatin States Drive Cryptic Transcription in AgingMammalian Stem Cells.
McCauley Brenna S et al.
A repressive chromatin state featuring trimethylated lysine 36 on histone H3 (H3K36me3) and DNA methylation suppresses cryptic transcription in embryonic stem cells. Cryptic transcription is elevated with age in yeast and nematodes, and reducing it extends yeast lifespan, though whether this occurs in mammals is unk... |
Metabolically controlled histone H4K5 acylation/acetylation ratiodrives BRD4 genomic distribution.
Gao M. et al.
In addition to acetylation, histones are modified by a series of competing longer-chain acylations. Most of these acylation marks are enriched and co-exist with acetylation on active gene regulatory elements. Their seemingly redundant functions hinder our understanding of histone acylations' specific roles. Here, by... |
Heterogeneity of neurons reprogrammed from spinal cord astrocytes by theproneural factors Ascl1 and Neurogenin2
Kempf J. et al.
Summary Astrocytes are a viable source for generating new neurons via direct conversion. However, little is known about the neurogenic cascades triggered in astrocytes from different regions of the CNS. Here, we examine the transcriptome induced by the proneural factors Ascl1 and Neurog2 in spinal cord-derived astro... |
Lasp1 regulates adherens junction dynamics and fibroblast transformationin destructive arthritis
Beckmann D. et al.
The LIM and SH3 domain protein 1 (Lasp1) was originally cloned from metastatic breast cancer and characterised as an adaptor molecule associated with tumourigenesis and cancer cell invasion. However, the regulation of Lasp1 and its function in the aggressive transformation of cells is unclear. Here we use integrativ... |
The SAM domain-containing protein 1 (SAMD1) acts as a repressivechromatin regulator at unmethylated CpG islands
Stielow B. et al.
CpG islands (CGIs) are key regulatory DNA elements at most promoters, but how they influence the chromatin status and transcription remains elusive. Here, we identify and characterize SAMD1 (SAM domain-containing protein 1) as an unmethylated CGI-binding protein. SAMD1 has an atypical winged-helix domain that direct... |
Waves of sumoylation support transcription dynamics during adipocytedifferentiation
Zhao, X. et al.
Tight control of gene expression networks required for adipose tissue formation and plasticity is essential for adaptation to energy needs and environmental cues. However, little is known about the mechanisms that orchestrate the dramatic transcriptional changes leading to adipocyte differentiation. We investigated ... |
Polycomb Repressive Complex 2 and KRYPTONITE regulate pathogen-inducedprogrammed cell death in Arabidopsis.
Dvořák Tomaštíková E. et al.
The Polycomb Repressive Complex 2 (PRC2) is well-known for its role in controlling developmental transitions by suppressing the premature expression of key developmental regulators. Previous work revealed that PRC2 also controls the onset of senescence, a form of developmental programmed cell death (PCD) in plants. ... |
An integrated multi-omics analysis identifies prognostic molecularsubtypes of non-muscle-invasive bladder cancer
Lindskrog Sia Viborg et al.
The molecular landscape in non-muscle-invasive bladder cancer (NMIBC) is characterized by large biological heterogeneity with variable clinical outcomes. Here, we perform an integrative multi-omics analysis of patients diagnosed with NMIBC (n = 834). Transcriptomic analysis identifies four classes (1, ... |
Loss of SETD1B results in the redistribution of genomic H3K4me3 in theoocyte
Hanna, C. W. et al.
Histone 3 lysine 4 trimethylation (H3K4me3) is an epigenetic mark found at gene promoters and CpG islands. H3K4me3 is essential for mammalian development, yet mechanisms underlying its genomic targeting are poorly understood. H3K4me3 methyltransferases SETD1B and MLL2 are essential for oogenesis. We investigated cha... |
VPRBP functions downstream of the androgen receptor and OGT to restrict p53 activation in prostate cancer
Poulose N. et al.
Androgen receptor (AR) is a major driver of prostate cancer (PCa) initiation and progression. O-GlcNAc transferase (OGT), the enzyme that catalyses the covalent addition of UDP-N-acetylglucosamine (UDP-GlcNAc) to serine and threonine residues of proteins, is often up-regulated in PCa with its expression correlated w... |
JAZF1, A Novel p400/TIP60/NuA4 Complex Member, Regulates H2A.ZAcetylation at Regulatory Regions.
Procida, Tara and Friedrich, Tobias and Jack, Antonia P M and Peritore,Martina and Bönisch, Clemens and Eberl, H Christian and Daus, Nadine andKletenkov, Konstantin and Nist, Andrea and Stiewe, Thorsten and Borggrefe,Tilman and Mann, Matthias and Bartk
Histone variants differ in amino acid sequence, expression timing and genomic localization sites from canonical histones and convey unique functions to eukaryotic cells. Their tightly controlled spatial and temporal deposition into specific chromatin regions is accomplished by dedicated chaperone and/or remodeling c... |
Epigenetic impairment and blunted transcriptional response to Mycobacteriumtuberculosis of alveolar macrophages from persons living with HIV
Correa-Macedo, W. et al.
Persons living with HIV (PLWH) are at increased risk of tuberculosis (TB). HIV-associated TB is often the result of recent infection with Mycobacterium tuberculosis (Mtb) followed by rapid progression to disease. Alveolar macrophages (AM) are the first cells of the innate immune system that engage Mtb, but how HIV a... |
Histone demethylase JMJD2B/KDM4B regulates transcriptional program viadistinctive epigenetic targets and protein interactors for the maintenanceof trophoblast stem cells.
Mak, Kylie Hin-Man et al.
Trophoblast stem cell (TSC) is crucial to the formation of placenta in mammals. Histone demethylase JMJD2 (also known as KDM4) family proteins have been previously shown to support self-renewal and differentiation of stem cells. However, their roles in the context of the trophoblast lineage remain unclear. Here, we ... |
Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture.
Yusufova, Nevin and Kloetgen, Andreas and Teater, Matt and Osunsade,Adewola and Camarillo, Jeannie M and Chin, Christopher R and Doane, AshleyS and Venters, Bryan J and Portillo-Ledesma, Stephanie and Conway, Josephand Phillip, Jude M and Elemento, Oli
Linker histone H1 proteins bind to nucleosomes and facilitate chromatin compaction, although their biological functions are poorly understood. Mutations in the genes that encode H1 isoforms B-E (H1B, H1C, H1D and H1E; also known as H1-5, H1-2, H1-3 and H1-4, respectively) are highly recurrent in B cell lymphoma... |
Multi-omic comparison of Alzheimer's variants in human ESC-derivedmicroglia reveals convergence at APOE.
Liu, Tongfei and Zhu, Bing and Liu, Yan and Zhang, Xiaoming and Yin, Junand Li, Xiaoguang and Jiang, LuLin and Hodges, Andrew P and Rosenthal, SaraBrin and Zhou, Lisa and Yancey, Joel and McQuade, Amanda and Blurton-Jones,Mathew and Tanzi, Rudolph E an
Variations in many genes linked to sporadic Alzheimer's disease (AD) show abundant expression in microglia, but relationships among these genes remain largely elusive. Here, we establish isogenic human ESC-derived microglia-like cell lines (hMGLs) harboring AD variants in CD33, INPP5D, SORL1, and TREM2 loci and cura... |
A genetic variant controls interferon-β gene expression in human myeloidcells by preventing C/EBP-β binding on a conserved enhancer.
Assouvie, Anaïs and Rotival, Maxime and Hamroune, Juliette and Busso,Didier and Romeo, Paul-Henri and Quintana-Murci, Lluis and Rousselet,Germain
Interferon β (IFN-β) is a cytokine that induces a global antiviral proteome, and regulates the adaptive immune response to infections and tumors. Its effects strongly depend on its level and timing of expression. Therefore, the transcription of its coding gene IFNB1 is strictly controlled. We have previous... |
Increased H3K4me3 methylation and decreased miR-7113-5p expression lead toenhanced Wnt/β-catenin signaling in immune cells from PTSD patientsleading to inflammatory phenotype.
Bam, Marpe and Yang, Xiaoming and Busbee, Brandon P and Aiello, Allison Eand Uddin, Monica and Ginsberg, Jay P and Galea, Sandro and Nagarkatti,Prakash S and Nagarkatti, Mitzi
BACKGROUND: Posttraumatic stress disorder (PTSD) is a psychiatric disorder accompanied by chronic peripheral inflammation. What triggers inflammation in PTSD is currently unclear. In the present study, we identified potential defects in signaling pathways in peripheral blood mononuclear cells (PBMCs) from individual... |
Trans- and cis-acting effects of Firre on epigenetic features of theinactive X chromosome.
Fang, He and Bonora, Giancarlo and Lewandowski, Jordan P and Thakur,Jitendra and Filippova, Galina N and Henikoff, Steven and Shendure, Jay andDuan, Zhijun and Rinn, John L and Deng, Xinxian and Noble, William S andDisteche, Christine M
Firre encodes a lncRNA involved in nuclear organization. Here, we show that Firre RNA expressed from the active X chromosome maintains histone H3K27me3 enrichment on the inactive X chromosome (Xi) in somatic cells. This trans-acting effect involves SUZ12, reflecting interactions between Firre RNA and components of t... |
The histone H2B ubiquitin ligase RNF40 is required for HER2-drivenmammary tumorigenesis.
Wegwitz, Florian and Prokakis, Evangelos and Pejkovska, Anastasija andKosinsky, Robyn Laura and Glatzel, Markus and Pantel, Klaus and Wikman,Harriet and Johnsen, Steven A
The HER2-positive breast cancer subtype (HER2-BC) displays a particularly aggressive behavior. Anti-HER2 therapies have significantly improved the survival of patients with HER2-BC. However, a large number of patients become refractory to current targeted therapies, necessitating the development of new treatment str... |
Polycomb Repressive Complex 2-mediated histone modification H3K27me3 isassociated with embryogenic potential in Norway spruce.
Nakamura, Miyuki and Batista, Rita A and Köhler, Claudia and Hennig, Lars
Epigenetic reprogramming during germ cell formation is essential to gain pluripotency and thus embryogenic potential. The histone modification H3K27me3, which is catalysed by the Polycomb repressive complex 2 (PRC2), regulates important developmental processes in both plants and animals, and defects in PRC2 componen... |
Age-associated cryptic transcription in mammalian stem cells is linked topermissive chromatin at cryptic promoters
McCauley B. S. et al.
Suppressing spurious cryptic transcription by a repressive intragenic chromatin state featuring trimethylated lysine 36 on histone H3 (H3K36me3) and DNA methylation is critical for maintaining self-renewal capacity in mouse embryonic stem cells. In yeast and nematodes, such cryptic transcription is elevated with age... |
RNF40 exerts stage-dependent functions in differentiating osteoblasts andis essential for bone cell crosstalk.
Najafova, Zeynab and Liu, Peng and Wegwitz, Florian and Ahmad, Mubashir andTamon, Liezel and Kosinsky, Robyn Laura and Xie, Wanhua and Johnsen, StevenA and Tuckermann, Jan
The role of histone ubiquitination in directing cell lineage specification is only poorly understood. Our previous work indicated a role of the histone 2B ubiquitin ligase RNF40 in controlling osteoblast differentiation in vitro. Here, we demonstrate that RNF40 has a stage-dependent function in controlling osteoblas... |
Epigenetic regulation of the lineage specificity of primary human dermallymphatic and blood vascular endothelial cells.
Tacconi, Carlotta and He, Yuliang and Ducoli, Luca and Detmar, Michael
Lymphatic and blood vascular endothelial cells (ECs) share several molecular and developmental features. However, these two cell types possess distinct phenotypic signatures, reflecting their different biological functions. Despite significant advances in elucidating how the specification of lymphatic and blood vasc... |
Combined treatment with CBP and BET inhibitors reverses inadvertentactivation of detrimental super enhancer programs in DIPG cells.
Wiese, M and Hamdan, FH and Kubiak, K and Diederichs, C and Gielen, GHand Nussbaumer, G and Carcaboso, AM and Hulleman, E and Johnsen, SA andKramm, CM
Diffuse intrinsic pontine gliomas (DIPG) are the most aggressive brain tumors in children with 5-year survival rates of only 2%. About 85% of all DIPG are characterized by a lysine-to-methionine substitution in histone 3, which leads to global H3K27 hypomethylation accompanied by H3K27 hyperacetylation. Hyperacetyla... |
Exploring the virulence gene interactome with CRISPR/dCas9 in the humanmalaria parasite.
Bryant, JM and Baumgarten, S and Dingli, F and Loew, D and Sinha, A andClaës, A and Preiser, PR and Dedon, PC and Scherf, A
Mutually exclusive expression of the var multigene family is key to immune evasion and pathogenesis in Plasmodium falciparum, but few factors have been shown to play a direct role. We adapted a CRISPR-based proteomics approach to identify novel factors associated with var genes in their natural chromatin context. Ca... |
Tissue-Specific In Vivo Biotin Chromatin Immunoprecipitation withSequencing in Zebrafish and Chicken
Lukoseviciute, Martyna and Ling, Irving T.C. and Senanayake, Upeka andCandido-Ferreira, Ivan and Taylor, Gunes and Williams, Ruth M. andSauka-Spengler, Tatjana
Chromatin immunoprecipitation with sequencing (ChIP-seq) has been instrumental in understanding transcription factor (TF) binding during gene regulation. ChIP-seq requires specific antibodies against desired TFs, which are not available for numerous species. Here, we describe a tissue-specific biotin ChIP-seq protoc... |
The gut microbiome switches mutant p53 from tumour-suppressive tooncogenic.
Kadosh, E and Snir-Alkalay, I and Venkatachalam, A and May, S and Lasry, Aand Elyada, E and Zinger, A and Shaham, M and Vaalani, G and Mernberger, Mand Stiewe, T and Pikarsky, E and Oren, M and Ben-Neriah, Y
Somatic mutations in p53, which inactivate the tumour-suppressor function of p53 and often confer oncogenic gain-of-function properties, are very common in cancer. Here we studied the effects of hotspot gain-of-function mutations in Trp53 (the gene that encodes p53 in mice) in mouse models of WNT-driven intestinal c... |
Egr2-guided histone H2B monoubiquitination is required for peripheral nervous system myelination.
Wüst HM, Wegener A, Fröb F, Hartwig AC, Wegwitz F, Kari V, Schimmel M, Tamm ER, Johnsen SA, Wegner M, Sock E
Schwann cells are the nerve ensheathing cells of the peripheral nervous system. Absence, loss and malfunction of Schwann cells or their myelin sheaths lead to peripheral neuropathies such as Charcot-Marie-Tooth disease in humans. During Schwann cell development and myelination chromatin is dramatically modified. How... |
Battle of the sex chromosomes: competition between X- and Y-chromosomeencoded proteins for partner interaction and chromatin occupancy drivesmulti-copy gene expression and evolution in muroid rodents.
Moretti, C and Blanco, M and Ialy-Radio, C and Serrentino, ME and Gobé,C and Friedman, R and Battail, C and Leduc, M and Ward, MA and Vaiman, Dand Tores, F and Cocquet, J
Transmission distorters (TDs) are genetic elements that favor their own transmission to the detriments of others. Slx/Slxl1 (Sycp3-like-X-linked and Slx-like1) and Sly (Sycp3-like-Y-linked) are TDs which have been co-amplified on the X and Y chromosomes of Mus species. They are involved in an intragenomic conflict i... |
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters hepatic polyunsaturated fatty acid metabolism and eicosanoid biosynthesis in female Sprague-Dawley rats.
Doskey CM, Fader KA, Nault R, Lydic T, Matthews J, Potter D, Sharratt B, Williams K, Zacharewski T
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a potent aryl hydrocarbon receptor (AhR) agonist that elicits a broad spectrum of dose-dependent hepatic effects including lipid accumulation, inflammation, and fibrosis. To determine the role of inflammatory lipid mediators in TCDD-mediated hepatotoxicity, eicosanoid me... |
Epigenetic priming by Dppa2 and 4 in pluripotency facilitates multi-lineage commitment.
Eckersley-Maslin MA, Parry A, Blotenburg M, Krueger C, Ito Y, Franklin VNR, Narita M, D'Santos CS, Reik W
How the epigenetic landscape is established in development is still being elucidated. Here, we uncover developmental pluripotency associated 2 and 4 (DPPA2/4) as epigenetic priming factors that establish a permissive epigenetic landscape at a subset of developmentally important bivalent promoters characterized by lo... |
Vulnerability of drug-resistant EML4-ALK rearranged lung cancer to transcriptional inhibition.
Paliouras AR, Buzzetti M, Shi L, Donaldson IJ, Magee P, Sahoo S, Leong HS, Fassan M, Carter M, Di Leva G, Krebs MG, Blackhall F, Lovly CM, Garofalo M
A subset of lung adenocarcinomas is driven by the EML4-ALK translocation. Even though ALK inhibitors in the clinic lead to excellent initial responses, acquired resistance to these inhibitors due to on-target mutations or parallel pathway alterations is a major clinical challenge. Exploring these mechanisms of resis... |
Removal of H2Aub1 by ubiquitin-specific proteases 12 and 13 is required for stable Polycomb-mediated gene repression in Arabidopsis.
Kralemann LEM, Liu S, Trejo-Arellano MS, Muñoz-Viana R, Köhler C, Hennig L
BACKGROUND: Stable gene repression is essential for normal growth and development. Polycomb repressive complexes 1 and 2 (PRC1&2) are involved in this process by establishing monoubiquitination of histone 2A (H2Aub1) and subsequent trimethylation of lysine 27 of histone 3 (H3K27me3). Previous work proposed that ... |
Delineating the early transcriptional specification of the mammalian trachea and esophagus.
Kuwahara A, Lewis AE, Coombes C, Leung FS, Percharde M, Bush JO
The genome-scale transcriptional programs that specify the mammalian trachea and esophagus are unknown. Though NKX2-1 and SOX2 are hypothesized to be co-repressive master regulators of tracheoesophageal fates, this is untested at a whole transcriptomic scale and their downstream networks remain unidentified. By comb... |
Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response.
Béguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, Venturutti L, Rivas MA, Calvo-Fernández MT, Gutierrez J, Camarillo JM, Takata K, Tarte K, Kelleher NL, Steidl C, Mason CE, Elemento O, Allis CD, Kleinstein SH, Melnick AM
Follicular lymphomas (FLs) are slow-growing, indolent tumors containing extensive follicular dendritic cell (FDC) networks and recurrent EZH2 gain-of-function mutations. Paradoxically, FLs originate from highly proliferative germinal center (GC) B cells with proliferation strictly dependent on interactions with T fo... |
H2A.Z is dispensable for both basal and activated transcription inpost-mitotic mouse muscles.
Belotti E. et al.
While the histone variant H2A.Z is known to be required for mitosis, it is also enriched in nucleosomes surrounding the transcription start site of active promoters, implicating H2A.Z in transcription. However, evidence obtained so far mainly rely on correlational data generated in actively dividing cells. We have e... |
Multi-omic analysis of gametogenesis reveals a novel signature at the promoters and distal enhancers of active genes.
Crespo M, Damont A, Blanco M, Lastrucci E, Kennani SE, Ialy-Radio C, Khattabi LE, Terrier S, Louwagie M, Kieffer-Jaquinod S, Hesse AM, Bruley C, Chantalat S, Govin J, Fenaille F, Battail C, Cocquet J, Pflieger D
Epigenetic regulation of gene expression is tightly controlled by the dynamic modification of histones by chemical groups, the diversity of which has largely expanded over the past decade with the discovery of lysine acylations, catalyzed from acyl-coenzymes A. We investigated the dynamics of lysine acetylation and ... |
Anti-adipogenic signals at the onset of obesity-related inflammation in white adipose tissue.
Caputo T, Tran VDT, Bararpour N, Winkler C, Aguileta G, Trang KB, Giordano Attianese GMP, Wilson A, Thomas A, Pagni M, Guex N, Desvergne B, Gilardi F
Chronic inflammation that affects primarily metabolic organs, such as white adipose tissue (WAT), is considered as a major cause of human obesity-associated co-morbidities. However, the molecular mechanisms initiating this inflammation in WAT are poorly understood. By combining transcriptomics, ChIP-seq and modeling... |
HDAC3 functions as a positive regulator in Notch signal transduction.
Ferrante F, Giaimo BD, Bartkuhn M, Zimmermann T, Close V, Mertens D, Nist A, Stiewe T, Meier-Soelch J, Kracht M, Just S, Klöble P, Oswald F, Borggrefe T
Aberrant Notch signaling plays a pivotal role in T-cell acute lymphoblastic leukemia (T-ALL) and chronic lymphocytic leukemia (CLL). Amplitude and duration of the Notch response is controlled by ubiquitin-dependent proteasomal degradation of the Notch1 intracellular domain (NICD1), a hallmark of the leukemogenic pro... |
Granulins Regulate Aging Kinetics in the Adult Zebrafish Telencephalon.
Zambusi A, Pelin Burhan Ö, Di Giaimo R, Schmid B, Ninkovic J
Granulins (GRN) are secreted factors that promote neuronal survival and regulate inflammation in various pathological conditions. However, their roles in physiological conditions in the brain remain poorly understood. To address this knowledge gap, we analysed the telencephalon in Grn-deficient zebrafish and identif... |
An inferred fitness consequence map of the rice genome.
Joly-Lopez Z, Platts AE, Gulko B, Choi JY, Groen SC, Zhong X, Siepel A, Purugganan MD
The extent to which sequence variation impacts plant fitness is poorly understood. High-resolution maps detailing the constraint acting on the genome, especially in regulatory sites, would be beneficial as functional annotation of noncoding sequences remains sparse. Here, we present a fitness consequence (fitCons) m... |
Dual-initiation promoters with intertwined canonical and TCT/TOP transcription start sites diversify transcript processing.
Nepal C, Hadzhiev Y, Balwierz P, Tarifeño-Saldivia E, Cardenas R, Wragg JW, Suzuki AM, Carninci P, Peers B, Lenhard B, Andersen JB, Müller F
Variations in transcription start site (TSS) selection reflect diversity of preinitiation complexes and can impact on post-transcriptional RNA fates. Most metazoan polymerase II-transcribed genes carry canonical initiation with pyrimidine/purine (YR) dinucleotide, while translation machinery-associated genes carry p... |
A comprehensive epigenomic analysis of phenotypically distinguishable, genetically identical female and male Daphnia pulex.
Kvist J, Athanàsio CG, Pfrender ME, Brown JB, Colbourne JK, Mirbahai L
BACKGROUND: Daphnia species reproduce by cyclic parthenogenesis involving both sexual and asexual reproduction. The sex of the offspring is environmentally determined and mediated via endocrine signalling by the mother. Interestingly, male and female Daphnia can be genetically identical, yet display large difference... |
Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq.
Kingsley NB, Kern C, Creppe C, Hales EN, Zhou H, Kalbfleisch TS, MacLeod JN, Petersen JL, Finno CJ, Bellone RR
One of the primary aims of the Functional Annotation of ANimal Genomes (FAANG) initiative is to characterize tissue-specific regulation within animal genomes. To this end, we used chromatin immunoprecipitation followed by sequencing (ChIP-Seq) to map four histone modifications (H3K4me1, H3K4me3, H3K27ac, and H3K27me... |
Combinatorial action of NF-Y and TALE at embryonic enhancers defines distinct gene expression programs during zygotic genome activation in zebrafish.
Stanney W, Ladam F, Donaldson IJ, Parsons TJ, Maehr R, Bobola N, Sagerström CG
Animal embryogenesis is initiated by maternal factors, but zygotic genome activation (ZGA) shifts regulatory control to the embryo during blastula stages. ZGA is thought to be mediated by maternally provided transcription factors (TFs), but few such TFs have been identified in vertebrates. Here we report that NF-Y a... |
Discovery of a new predominant cytosine DNA modification that is linked to gene expression in malaria parasites.
Hammam E, Ananda G, Sinha A, Scheidig-Benatar C, Bohec M, Preiser PR, Dedon PC, Scherf A, Vembar SS
DNA cytosine modifications are key epigenetic regulators of cellular processes in mammalian cells, with their misregulation leading to varied disease states. In the human malaria parasite Plasmodium falciparum, a unicellular eukaryotic pathogen, little is known about the predominant cytosine modifications, cytosine ... |
A stress-responsive enhancer induces dynamic drug resistance in acute myeloid leukemia.
Williams MS, Amaral FM, Simeoni F, Somervaille TC
The drug efflux pump ABCB1 is a key driver of chemoresistance, and high expression predicts for treatment failure in acute myeloid leukemia (AML). In this study, we identified and functionally validated the network of enhancers that controls expression of ABCB1. We show that exposure of leukemia cells to daunorubici... |
Distinct CoREST complexes act in a cell-type-specific manner.
Mačinković I, Theofel I, Hundertmark T, Kovač K, Awe S, Lenz J, Forné I, Lamp B, Nist A, Imhof A, Stiewe T, Renkawitz-Pohl R, Rathke C, Brehm A
CoREST has been identified as a subunit of several protein complexes that generate transcriptionally repressive chromatin structures during development. However, a comprehensive analysis of the CoREST interactome has not been carried out. We use proteomic approaches to define the interactomes of two dCoREST isoforms... |
Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses.
Timofeev O, Klimovich B, Schneikert J, Wanzel M, Pavlakis E, Noll J, Mutlu S, Elmshäuser S, Nist A, Mernberger M, Lamp B, Wenig U, Brobeil A, Gattenlöhner S, Köhler K, Stiewe T
Engineered p53 mutant mice are valuable tools for delineating p53 functions in tumor suppression and cancer therapy. Here, we have introduced the R178E mutation into the Trp53 gene of mice to specifically ablate the cooperative nature of p53 DNA binding. Trp53 mice show no detectable target gene regulation and, at f... |
EOMES interacts with RUNX3 and BRG1 to promote innate memory cell formation through epigenetic reprogramming.
Istaces N, Splittgerber M, Lima Silva V, Nguyen M, Thomas S, Le A, Achouri Y, Calonne E, Defrance M, Fuks F, Goriely S, Azouz A
Memory CD8 T cells have the ability to provide lifelong immunity against pathogens. Although memory features generally arise after challenge with a foreign antigen, naïve CD8 single positive (SP) thymocytes may acquire phenotypic and functional characteristics of memory cells in response to cytokines such as in... |
Development and epigenetic plasticity of murine Müller glia.
Dvoriantchikova G, Seemungal RJ, Ivanov D
The ability to regenerate the entire retina and restore lost sight after injury is found in some species and relies mostly on the epigenetic plasticity of Müller glia. To understand the role of mammalian Müller glia as a source of progenitors for retinal regeneration, we investigated changes in gene expres... |
ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells.
Sen M, Wang X, Hamdan FH, Rapp J, Eggert J, Kosinsky RL, Wegwitz F, Kutschat AP, Younesi FS, Gaedcke J, Grade M, Hessmann E, Papantonis A, Strӧbel P, Johnsen SA
BACKGROUND: ARID1A (AT-rich interactive domain-containing protein 1A) is a subunit of the BAF chromatin remodeling complex and plays roles in transcriptional regulation and DNA damage response. Mutations in ARID1A that lead to inactivation or loss of expression are frequent and widespread across many cancer typ... |
Guidelines for optimized gene knockout using CRISPR/Cas9
Campenhout CV et al.
CRISPR/Cas9 technology has evolved as the most powerful approach to generate genetic models both for fundamental and preclinical research. Despite its apparent simplicity, the outcome of a genome-editing experiment can be substantially impacted by technical parameters and biological considerations. Here, we present ... |
BRCA1 mutations attenuate super-enhancer function and chromatin looping in haploinsufficient human breast epithelial cells.
Zhang X, Wang Y, Chiang HC, Hsieh YP, Lu C, Park BH, Jatoi I, Jin VX, Hu Y, Li R
BACKGROUND: BRCA1-associated breast cancer originates from luminal progenitor cells. BRCA1 functions in multiple biological processes, including double-strand break repair, replication stress suppression, transcriptional regulation, and chromatin reorganization. While non-malignant cells carrying cancer-predisposing... |
Point mutations in the PDX1 transactivation domain impair human β-cell development and function.
Wang X, Sterr M, Ansarullah , Burtscher I, Böttcher A, Beckenbauer J, Siehler J, Meitinger T, Häring HU, Staiger H, Cernilogar FM, Schotta G, Irmler M, Beckers J, Wright CVE, Bakhti M, Lickert H
OBJECTIVE: Hundreds of missense mutations in the coding region of PDX1 exist; however, if these mutations predispose to diabetes mellitus is unknown. METHODS: In this study, we screened a large cohort of subjects with increased risk for diabetes and identified two subjects with impaired glucose tolerance carrying co... |
A critical regulator of Bcl2 revealed by systematic transcript discovery of lncRNAs associated with T-cell differentiation.
Saadi W, Kermezli Y, Dao LTM, Mathieu E, Santiago-Algarra D, Manosalva I, Torres M, Belhocine M, Pradel L, Loriod B, Aribi M, Puthier D, Spicuglia S
Normal T-cell differentiation requires a complex regulatory network which supports a series of maturation steps, including lineage commitment, T-cell receptor (TCR) gene rearrangement, and thymic positive and negative selection. However, the underlying molecular mechanisms are difficult to assess due to limited T-ce... |
Transcriptome-wide dynamics of extensive m6A mRNA methylation during Plasmodium falciparum blood-stage development
Sebastian Baumgarten, Jessica M. Bryant, Ameya Sinha, Thibaud Reyser, Peter R. Preiser, Peter C. Dedon, Artur Scherf
Malaria pathogenesis results from the asexual replication of Plasmodium falciparum within human red blood cells, which relies on a precisely timed cascade of gene expression over a 48-hour life cycle. Although substantial post-transcriptional regulation of this hardwired program has been observed, it remains unclear... |
The epigenetic basis for the impaired ability of adult murine retinal pigment epithelium cells to regenerate retinal tissue.
Dvoriantchikova G, Seemungal RJ, Ivanov D
The epigenetic plasticity of amphibian retinal pigment epithelium (RPE) allows them to regenerate the entire retina, a trait known to be absent in mammals. In this study, we investigated the epigenetic plasticity of adult murine RPE to identify possible mechanisms that prevent mammalian RPE from regenerating retinal... |
NF-κB p65 dimerization and DNA-binding is important for inflammatory gene expression.
Riedlinger T, Liefke R, Meier-Soelch J, Jurida L, Nist A, Stiewe T, Kracht M, Schmitz ML
Increasing evidence shows that many transcription factors execute important biologic functions independent from their DNA-binding capacity. The NF-κB p65 (RELA) subunit is a central regulator of innate immunity. Here, we investigated the relative functional contribution of p65 DNA-binding and dimerization in p... |
Differential regulation of RNA polymerase III genes during liver regeneration.
Yeganeh M, Praz V, Carmeli C, Villeneuve D, Rib L, Guex N, Herr W, Delorenzi M, Hernandez N,
Mouse liver regeneration after partial hepatectomy involves cells in the remaining tissue synchronously entering the cell division cycle. We have used this system and H3K4me3, Pol II and Pol III profiling to characterize adaptations in Pol III transcription. Our results broadly define a class of genes close to H3K4m... |
CBX7 Induces Self-Renewal of Human Normal and Malignant Hematopoietic Stem and Progenitor Cells by Canonical and Non-canonical Interactions.
Jung J, Buisman SC, Weersing E, Dethmers-Ausema A, Zwart E, Schepers H, Dekker MR, Lazare SS, Hammerl F, Skokova Y, Kooistra SM, Klauke K, Poot RA, Bystrykh LV, de Haan G
In this study, we demonstrate that, among all five CBX Polycomb proteins, only CBX7 possesses the ability to control self-renewal of human hematopoietic stem and progenitor cells (HSPCs). Xenotransplantation of CBX7-overexpressing HSPCs resulted in increased multi-lineage long-term engraftment and myelopoiesis. ... |
Fluorescence-Activated Cell Sorting-Based Isolation and Characterization of Neural Stem Cells from the Adult Zebrafish Telencephalon.
Di Giaimo R, Aschenbroich S, Ninkovic J
Adult mammalian brain, including humans, has rather limited addition of new neurons and poor regenerative capacity. In contrast, neural stem cells (NSC) with glial identity and neurogenesis are highly abundant throughout the adult zebrafish brain. Importantly, the activation of NSC and production of new neurons in r... |
DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network.
Hamdan FH, Johnsen SA
Molecular subtyping of cancer offers tremendous promise for the optimization of a precision oncology approach to anticancer therapy. Recent advances in pancreatic cancer research uncovered various molecular subtypes with tumors expressing a squamous/basal-like gene expression signature displaying a worse prognosis. ... |
The Aryl Hydrocarbon Receptor Pathway Defines the Time Frame for Restorative Neurogenesis.
Di Giaimo R, Durovic T, Barquin P, Kociaj A, Lepko T, Aschenbroich S, Breunig CT, Irmler M, Cernilogar FM, Schotta G, Barbosa JS, Trümbach D, Baumgart EV, Neuner AM, Beckers J, Wurst W, Stricker SH, Ninkovic J
Zebrafish have a high capacity to replace lost neurons after brain injury. New neurons involved in repair are generated by a specific set of glial cells, known as ependymoglial cells. We analyze changes in the transcriptome of ependymoglial cells and their progeny after injury to infer the molecular pathways governi... |
Genome-wide identification of RETINOBLASTOMA RELATED 1 binding sites in Arabidopsis reveals novel DNA damage regulators.
Bouyer D, Heese M, Chen P, Harashima H, Roudier F, Grüttner C, Schnittger A
Retinoblastoma (pRb) is a multifunctional regulator, which was likely present in the last common ancestor of all eukaryotes. The Arabidopsis pRb homolog RETINOBLASTOMA RELATED 1 (RBR1), similar to its animal counterparts, controls not only cell proliferation but is also implicated in developmental decisions, stress ... |
SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States
Cossec Jack-Christophe, Theurillat Ilan, Chica Claudia, Búa Aguín Sabela, Gaume Xavier, Andrieux Alexandra, Iturbide Ane, Jouvion Gregory, Li Han, Bossis Guillaume, Seeler Jacob-Sebastian, Torres-Padilla Maria-Elena, Dejean Anne
Understanding general principles that safeguard cellular identity should reveal critical insights into common mechanisms underlying specification of varied cell types. Here, we show that SUMO modification acts to stabilize cell fate in a variety of contexts. Hyposumoylation enhances pluripotency reprogramming in vit... |
Caenorhabditis elegans sperm carry a histone-based epigenetic memory of both spermatogenesis and oogenesis.
Tabuchi TM, Rechtsteiner A, Jeffers TE, Egelhofer TA, Murphy CT, Strome S
Paternal contributions to epigenetic inheritance are not well understood. Paternal contributions via marked nucleosomes are particularly understudied, in part because sperm in some organisms replace the majority of nucleosome packaging with protamine packaging. Here we report that in Caenorhabditis elegans sperm, th... |
PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex.
Link S, Spitzer RMM, Sana M, Torrado M, Völker-Albert MC, Keilhauer EC, Burgold T, Pünzeler S, Low JKK, Lindström I, Nist A, Regnard C, Stiewe T, Hendrich B, Imhof A, Mann M, Mackay JP, Bartkuhn M, Hake SB
Chromatin structure and function is regulated by reader proteins recognizing histone modifications and/or histone variants. We recently identified that PWWP2A tightly binds to H2A.Z-containing nucleosomes and is involved in mitotic progression and cranial-facial development. Here, using in vitro assays, we show that... |
Convergent evolution of complex genomic rearrangements in two fungal meiotic drive elements.
Svedberg J, Hosseini S, Chen J, Vogan AA, Mozgova I, Hennig L, Manitchotpisit P, Abusharekh A, Hammond TM, Lascoux M, Johannesson H
Meiotic drive is widespread in nature. The conflict it generates is expected to be an important motor for evolutionary change and innovation. In this study, we investigated the genomic consequences of two large multi-gene meiotic drive elements, Sk-2 and Sk-3, found in the filamentous ascomycete Neurospora intermedi... |
Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones.
de Boer B, Prick J, Pruis MG, Keane P, Imperato MR, Jaques J, Brouwers-Vos AZ, Hogeling SM, Woolthuis CM, Nijk MT, Diepstra A, Wandinger S, Versele M, Attar RM, Cockerill PN, Huls G, Vellenga E, Mulder AB, Bonifer C, Schuringa JJ
Intra-tumor heterogeneity caused by clonal evolution is a major problem in cancer treatment. To address this problem, we performed label-free quantitative proteomics on primary acute myeloid leukemia (AML) samples. We identified 50 leukemia-enriched plasma membrane proteins enabling the prospective isolation of gene... |
Genomic Location of PRMT6-Dependent H3R2 Methylation Is Linked to the Transcriptional Outcome of Associated Genes.
Bouchard C, Sahu P, Meixner M, Nötzold RR, Rust MB, Kremmer E, Feederle R, Hart-Smith G, Finkernagel F, Bartkuhn M, Savai Pullamsetti S, Nist A, Stiewe T, Philipsen S, Bauer UM
Protein arginine methyltransferase 6 (PRMT6) catalyzes asymmetric dimethylation of histone H3 at arginine 2 (H3R2me2a). This mark has been reported to associate with silent genes. Here, we use a cell model of neural differentiation, which upon PRMT6 knockout exhibits proliferation and differentiation defects. S... |
Enhancer-driven transcriptional regulation is a potential key determinant for human visceral and subcutaneous adipocytes.
Liefke R, Bokelmann K, Ghadimi BM, Dango S
Obesity is characterized by the excess of body fat leading to impaired health. Abdominal fat is particularly harmful and is associated with cardiovascular and metabolic diseases and cancer. In contrast, subcutaneous fat is generally considered less detrimental. The mechanisms that establish the cellular characterist... |
Episomal HBV persistence within transcribed host nuclear chromatin compartments involves HBx.
Hensel KO, Cantner F, Bangert F, Wirth S, Postberg J
BACKGROUND: In hepatocyte nuclei, hepatitis B virus (HBV) genomes occur episomally as covalently closed circular DNA (cccDNA). The HBV X protein (HBx) is required to initiate and maintain HBV replication. The functional nuclear localization of cccDNA and HBx remains unexplored. RESULTS: To identify virus-host genome... |
Genome-wide rules of nucleosome phasing
Sandro Baldi, Dhawal S. Jain1, Lisa Harpprecht1, Angelika Zabel1, Marion Scheibe, Falk Butter, Tobias Straub and Peter B. Becker
Regular successions of positioned nucleosomes – phased nucleosome arrays (PNAs) – are predominantly known from transcriptional start sites (TSS). It is unclear whether PNAs occur elsewhere in the genome. To generate a comprehensive inventory of PNAs for Drosophila, we applied spectral analysis to nucleos... |
Insulin promoter in human pancreatic β cells contacts diabetes susceptibility loci and regulates genes affecting insulin metabolism.
Jian X, Felsenfeld G
Both type 1 and type 2 diabetes involve a complex interplay between genetic, epigenetic, and environmental factors. Our laboratory has been interested in the physical interactions, in nuclei of human pancreatic β cells, between the insulin ( gene and other genes that are involved in insulin metabolism. We have ... |
Modulation of gene transcription and epigenetics of colon carcinoma cells by bacterial membrane vesicles.
Vdovikova S, Gilfillan S, Wang S, Dongre M, Wai SN, Hurtado A
Interactions between bacteria and colon cancer cells influence the transcription of the host cell. Yet is it undetermined whether the bacteria itself or the communication between the host and bacteria is responsible for the genomic changes in the eukaryotic cell. Now, we have investigated the genomic and epigenetic ... |
Combined cistrome and transcriptome analysis of SKI in AML cells identifies SKI as a co-repressor for RUNX1.
Feld C, Sahu P, Frech M, Finkernagel F, Nist A, Stiewe T, Bauer UM, Neubauer A
SKI is a transcriptional co-regulator and overexpressed in various human tumors, for example in acute myeloid leukemia (AML). SKI contributes to the origin and maintenance of the leukemic phenotype. Here, we use ChIP-seq and RNA-seq analysis to identify the epigenetic alterations induced by SKI overexpression in AML... |
A long range distal enhancer controls temporal fine-tuning of PAX6 expression in neuronal precursors.
Lacomme M, Medevielle F, Bourbon HM, Thierion E, Kleinjan DJ, Roussat M, Pituello F, Bel-Vialar S
Proper embryonic development relies on a tight control of spatial and temporal gene expression profiles in a highly regulated manner. One good example is the ON/OFF switching of the transcription factor PAX6 that governs important steps of neurogenesis. In the neural tube PAX6 expression is initiated in neural proge... |
HDAC1 and HDAC2 Modulate TGF-β Signaling during Endothelial-to-Hematopoietic Transition.
Thambyrajah R, Fadlullah MZH, Proffitt M, Patel R, Cowley SM, Kouskoff V, Lacaud G
The first hematopoietic stem and progenitor cells are generated during development from hemogenic endothelium (HE) through trans-differentiation. The molecular mechanisms underlying this endothelial-to-hematopoietic transition (EHT) remain poorly understood. Here, we explored the role of the epigenetic regulators HD... |
EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma.
Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, Iniguez AB, Bernard X, Masamsetti VP, Roston S, Lawlor ER, Toretsky JA, Stegmaier K, Lessnick SL, Chen Y, Bishop AJR
Ewing sarcoma is an aggressive paediatric cancer of the bone and soft tissue. It results from a chromosomal translocation, predominantly t(11;22)(q24:q12), that fuses the N-terminal transactivation domain of the constitutively expressed EWSR1 protein with the C-terminal DNA binding domain of the rarely expressed FLI... |
A Specific PfEMP1 Is Expressed in P. falciparum Sporozoites and Plays a Role in Hepatocyte Infection.
Zanghì G, Vembar SS, Baumgarten S, Ding S, Guizetti J, Bryant JM, Mattei D, Jensen ATR, Rénia L, Goh YS, Sauerwein R, Hermsen CC, Franetich JF, Bordessoulles M, Silvie O, Soulard V, Scatton O, Chen P, Mecheri S, Mazier D, Scherf A
Heterochromatin plays a central role in the process of immune evasion, pathogenesis, and transmission of the malaria parasite Plasmodium falciparum during blood stage infection. Here, we use ChIP sequencing to demonstrate that sporozoites from mosquito salivary glands expand heterochromatin at subtelomeric regions t... |
Genome-wide analysis of PDX1 target genes in human pancreatic progenitors.
Wang X, Sterr M, Burtscher I, Chen S, Hieronimus A, Machicao F, Staiger H, Häring HU, Lederer G, Meitinger T, Cernilogar FM, Schotta G, Irmler M, Beckers J, Hrabě de Angelis M, Ray M, Wright CVE, Bakhti M, Lickert H
OBJECTIVE: Homozygous loss-of-function mutations in the gene coding for the homeobox transcription factor (TF) PDX1 leads to pancreatic agenesis, whereas heterozygous mutations can cause Maturity-Onset Diabetes of the Young 4 (MODY4). Although the function of Pdx1 is well studied in pre-clinical models during insuli... |
A multi-omics study of the grapevine-downy mildew (Plasmopara viticola) pathosystem unveils a complex protein coding- and noncoding-based arms race during infection.
Brilli M, Asquini E, Moser M, Bianchedi PL, Perazzolli M, Si-Ammour A
Fungicides are applied intensively to prevent downy mildew infections of grapevines (Vitis vinifera) with high impact on the environment. In order to develop alternative strategies we sequenced the genome of the oomycete pathogen Plasmopara viticola causing this disease. We show that it derives from a Phytophthora-l... |
BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis.
Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier JC, Mailhot-Léonard F, Ahmed E, Belle J, Besla R, Mazer B, King IL, Nijnik A, Robbins CS, Barreiro LB, Divangahi M
The dogma that adaptive immunity is the only arm of the immune response with memory capacity has been recently challenged by several studies demonstrating evidence for memory-like innate immune training. However, the underlying mechanisms and location for generating such innate memory responses in vivo rem... |
MLL2 conveys transcription-independent H3K4 trimethylation in oocytes
Hanna C.W. et al.
Histone 3 K4 trimethylation (depositing H3K4me3 marks) is typically associated with active promoters yet paradoxically occurs at untranscribed domains. Research to delineate the mechanisms of targeting H3K4 methyltransferases is ongoing. The oocyte provides an attractive system to investigate these mechanisms, becau... |
Functional dissection of Drosophila melanogaster SUUR protein influence on H3K27me3 profile
Posukh O. V. et al.
Background
In eukaryotes, heterochromatin replicates late in S phase of the cell cycle and contains specific covalent modifications of histones. SuUR mutation found in Drosophila makes heterochromatin replicate earlier than in wild type and reduces the level of repressive histone modifications. SUUR protein was s... |
An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells
Berrens R.V. et al.
Erasure of DNA methylation and repressive chromatin marks in the mammalian germline leads to risk of transcriptional activation of transposable elements (TEs). Here, we used mouse embryonic stem cells (ESCs) to identify an endosiRNA-based mechanism involved in suppression of TE transcription. In ESCs with DNA demeth... |
Epigenome profiling and editing of neocortical progenitor cells during development
Albert M. et al.
The generation of neocortical neurons from neural progenitor cells (NPCs) is primarily controlled by transcription factors binding to DNA in the context of chromatin. To understand the complex layer of regulation that orchestrates different NPC types from the same DNA sequence, epigenome maps with cell type resoluti... |
Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles
Marincevic-Zuniga Y. et al.
BACKGROUND:
Structural chromosomal rearrangements that lead to expressed fusion genes are a hallmark of acute lymphoblastic leukemia (ALL). In this study, we performed transcriptome sequencing of 134 primary ALL patient samples to comprehensively detect fusion transcripts.
METHODS:
We combined fusion gene detec... |
Dynamics of RNA Polymerase II Pausing and Bivalent Histone H3 Methylation during Neuronal Differentiation in Brain Development
Liu J. et al.
During cellular differentiation, genes important for differentiation are expected to be silent in stem/progenitor cells yet can be readily activated. RNA polymerase II (Pol II) pausing and bivalent chromatin marks are two paradigms suited for establishing such a poised state of gene expression; however, their specif... |
Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation
Pünzeler S. et al.
Replacement of canonical histones with specialized histone variants promotes altering of chromatin structure and function. The essential histone variant H2A.Z affects various DNA-based processes via poorly understood mechanisms. Here, we determine the comprehensive interactome of H2A.Z and identify PWWP2A as a novel... |
Krox20 hindbrain regulation incorporates multiple modes of cooperation between cis-acting elements
Thierion E. et al.
Developmental genes can harbour multiple transcriptional enhancers that act simultaneously or in succession to achieve robust and precise spatiotemporal expression. However, the mechanisms underlying cooperation between cis-acting elements are poorly documented, notably in vertebrates. The mouse gene Krox20 encodes ... |
Ectopic application of the repressive histone modification H3K9me2 establishes post-zygotic reproductive isolation in Arabidopsis thaliana
Jiang H. et al.
Hybrid seed lethality as a consequence of interspecies or interploidy hybridizations is a major mechanism of reproductive isolation in plants. This mechanism is manifested in the endosperm, a dosage-sensitive tissue supporting embryo growth. Deregulated expression of imprinted genes such as ADMETOS (ADM) underpin th... |
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-elicited effects on bile acid homeostasis: Alterations in biosynthesis, enterohepatic circulation, and microbial metabolism.
Fader K. et al.
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is a persistent environmental contaminant which elicits hepatotoxicity through activation of the aryl hydrocarbon receptor (AhR). Male C57BL/6 mice orally gavaged with TCDD (0.01-30 µg/kg) every 4 days for 28 days exhibited bile duct proliferation and perichola... |
Platelet function is modified by common sequence variation in megakaryocyte super enhancers
Petersen R. et al.
Linking non-coding genetic variants associated with the risk of diseases or disease-relevant traits to target genes is a crucial step to realize GWAS potential in the introduction of precision medicine. Here we set out to determine the mechanisms underpinning variant association with platelet quantitative traits usi... |
CRISPR/Cas9 Genome Editing Reveals That the Intron Is Not Essential for var2csa Gene Activation or Silencing in Plasmodium falciparum
Bryant J.M. et al.
Plasmodium falciparum relies on monoallelic expression of 1 of 60 var virulence genes for antigenic variation and host immune evasion. Each var gene contains a conserved intron which has been implicated in previous studies in both activation and repression of transcription via several epigenetic mechanisms, includin... |
Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)- and aryl hydrocarbon receptor repressor (AHRR)-binding sites in human breast cancer cells
Sunny Y. Yang, Shaimaa Ahmed, Somisetty V. Satheesh, Jason Matthews
The aryl hydrocarbon receptor (AHR) mediates the toxic actions of environmental contaminants, such as 2,3,7,8-tetrachlorodibenzo-ρ-dioxin (TCDD), and also plays roles in vascular development, the immune response, and cell cycle regulation. The AHR repressor (AHRR) is an AHR-regulated gene and a negative regulato... |
RNA Polymerase III Subunit POLR3G Regulates Specific Subsets of PolyA(+) and SmallRNA Transcriptomes and Splicing in Human Pluripotent Stem Cells.
Lund R.J. et al.
POLR3G is expressed at high levels in human pluripotent stem cells (hPSCs) and is required for maintenance of stem cell state through mechanisms not known in detail. To explore how POLR3G regulates stem cell state, we carried out deep-sequencing analysis of polyA+ and smallRNA transcriptomes present in hPSCs an... |
Characterization of the Polycomb-Group Mark H3K27me3 in Unicellular Algae
Mikulski P. et al.
Polycomb Group (PcG) proteins mediate chromatin repression in plants and animals by catalyzing H3K27 methylation and H2AK118/119 mono-ubiquitination through the activity of the Polycomb repressive complex 2 (PRC2) and PRC1, respectively. PcG proteins were extensively studied in higher plants, but their function and ... |
Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions
Frank-Bertoncelj M, Trenkmann M, Klein K, Karouzakis E, Rehrauer H, Bratus A, Kolling C, Armaka M, Filer A, Michel BA, Gay RE, Buckley CD, Kollias G, Gay S, Ospelt C
A number of human diseases, such as arthritis and atherosclerosis, include characteristic pathology in specific anatomical locations. Here we show transcriptomic differences in synovial fibroblasts from different joint locations and that HOX gene signatures reflect the joint-specific origins of mouse and human synov... |
First landscape of binding to chromosomes for a domesticated mariner transposase in the human genome: diversity of genomic targets of SETMAR isoforms in two colorectal cell lines
Antoine-Lorquin A. et al.
Setmar is a 3-exons gene coding a SET domain fused to a Hsmar1 transposase. Its different transcripts theoretically encode 8 isoforms with SET moieties differently spliced. In vitro, the largest isoform binds specifically to Hsmar1 DNA ends and with no specificity to DNA when it is associated with hPso4. In colon ce... |
Crebbp loss cooperates with Bcl2 over-expression to promote lymphoma in mice
Idoia García-Ramírez, Saber Tadros, Inés González-Herrero, Alberto Martín-Lorenzo, Guillermo Rodríguez-Hernández, Dalia Moore, Lucía Ruiz-Roca, Oscar Blanco, Diego Alonso-López, Javier De Las Rivas, Keenan Hartert, Romain Duval, David Klinkebiel, Martin B
CREBBP is targeted by inactivating mutations in follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL). Here, we provide evidence from transgenic mouse models that Crebbp deletion results in deficits in B-cell development and can cooperate with Bcl2 over-expression to promote B-cell lymphoma. Through tra... |
Aorta macrophage inflammatory and epigenetic changes in a murine model of obstructive sleep apnea: Potential role of CD36.
Cortese R. et al.
Obstructive sleep apnea (OSA) affects 8-10% of the population, is characterized by chronic intermittent hypoxia (CIH), and causally associates with cardiovascular morbidities. In CIH-exposed mice, closely mimicking the chronicity of human OSA, increased accumulation and proliferation of pro-inflammatory metabolic M1... |
Intestinal NCoR1, a regulator of epithelial cell maturation, controls neonatal hyperbilirubinemia
Chen S. et al.
Severe neonatal hyperbilirubinemia (SNH) and the onset of bilirubin encephalopathy and kernicterus result in part from delayed expression of UDP-glucuronosyltransferase 1A1 (UGT1A1) and the inability to metabolize bilirubin. Although there is a good understanding of the early events after birth that lead to the rapi... |
The Drosophila speciation factor HMR localizes to genomic insulator sites
Gerland T.A. et al.
Hybrid incompatibility between Drosophila melanogaster and D. simulans is caused by a lethal interaction of the proteins encoded by the Hmr and Lhr genes. In D. melanogaster the loss of HMR results in mitotic defects, an increase in transcription of transposable elements and a deregulation of heterochromatic genes. ... |
Applying the INTACT method to purify endosperm nuclei and to generate parental-specific epigenome profiles.
Moreno-Romero J. et al.
The early endosperm tissue of dicot species is very difficult to isolate by manual dissection. This protocol details how to apply the INTACT (isolation of nuclei tagged in specific cell types) system for isolating early endosperm nuclei of Arabidopsis at high purity and how to generate parental-specific epigenome pr... |
BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire
Najafova Z. et al.
Proper temporal epigenetic regulation of gene expression is essential for cell fate determination and tissue development. The Bromodomain-containing Protein-4 (BRD4) was previously shown to control the transcription of defined subsets of genes in various cell systems. In this study we examined the role of BRD4 in pr... |
CTCF modulates Estrogen Receptor function through specific chromatin and nuclear matrix interactions
Fiorito E. et al.
Enhancer regions and transcription start sites of estrogen-target regulated genes are connected by means of Estrogen Receptor long-range chromatin interactions. Yet, the complete molecular mechanisms controlling the transcriptional output of engaged enhancers and subsequent activation of coding genes remain elusive.... |
PionX sites mark the X chromosome for dosage compensation
Villa R et al.
The rules defining which small fraction of related DNA sequences can be selectively bound by a transcription factor are poorly understood. One of the most challenging tasks in DNA recognition is posed by dosage compensation systems that require the distinction between sex chromosomes and autosomes. In Drosophila mel... |
reChIP-seq reveals widespread bivalency of H3K4me3 and H3K27me3 in CD4(+) memory T cells
Kinkley S et al.
The combinatorial action of co-localizing chromatin modifications and regulators determines chromatin structure and function. However, identifying co-localizing chromatin features in a high-throughput manner remains a technical challenge. Here we describe a novel reChIP-seq approach and tailored bioinformatic analys... |
Deletion of Polycomb Repressive Complex 2 From Mouse Intestine Causes Loss of Stem Cells
Koppens MA et al.
BACKGROUND & AIMS:
The polycomb repressive complex 2 (PRC2) regulates differentiation by contributing to repression of gene expression and thereby stabilizing the fate of stem cells and their progeny. PRC2 helps to maintain adult stem cell populations, but little is known about its functions in intestinal stem ... |
Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells
von Meyenn F et al.
Global demethylation is part of a conserved program of epigenetic reprogramming to naive pluripotency. The transition from primed hypermethylated embryonic stem cells (ESCs) to naive hypomethylated ones (serum-to-2i) is a valuable model system for epigenetic reprogramming. We present a mathematical model, which accu... |
Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm
Moreno-Romero J et al.
Parental genomes in the endosperm are marked by differential DNA methylation and are therefore epigenetically distinct. This epigenetic asymmetry is established in the gametes and maintained after fertilization by unknown mechanisms. In this manuscript, we have addressed the key question whether parentally inherited... |